iTRAQ Proteomics
Gabriel Mateus Bernardo Harrington
https://h-mateus.github.io/presentations/
2020-10-01
History
- Developed in 2004 by Applied Biosystems Incorporation
- Abbreviation: isobaric tag for relative and absolute quantitation
- Method for determining the amount of proteins from different samples in a single mass spec run
iTRAQ reagents
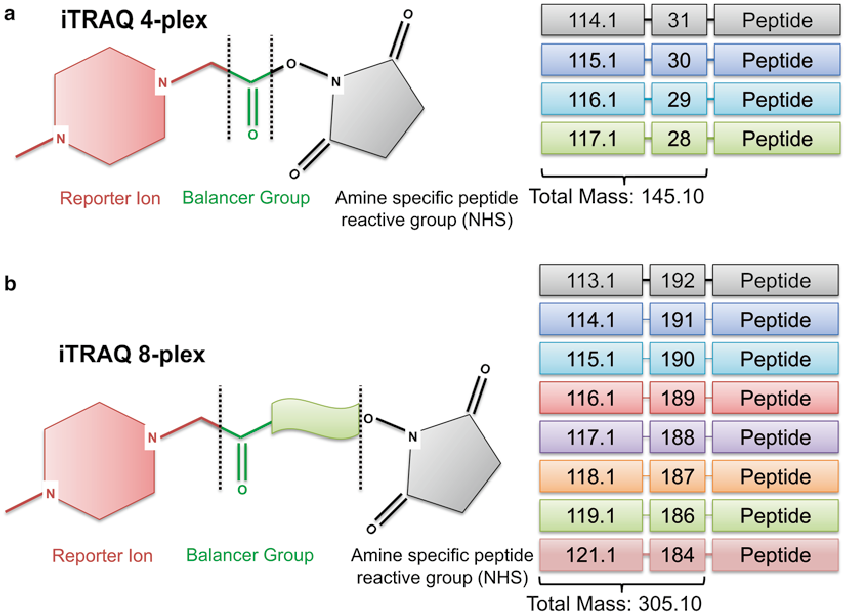
Details
- Consist of a unique charged reporter group, a peptide reactive group and a neutral balance group
- The peptide reactive group covalently links an isobaric tag to the lysine side chain and N-terminus group of a peptide
- The balance group ensures labelled peptide displays the same mass to maintain an overall mass of 145 Da for 4-plex and 305 Da for 8-plex
iTRAQ workflow
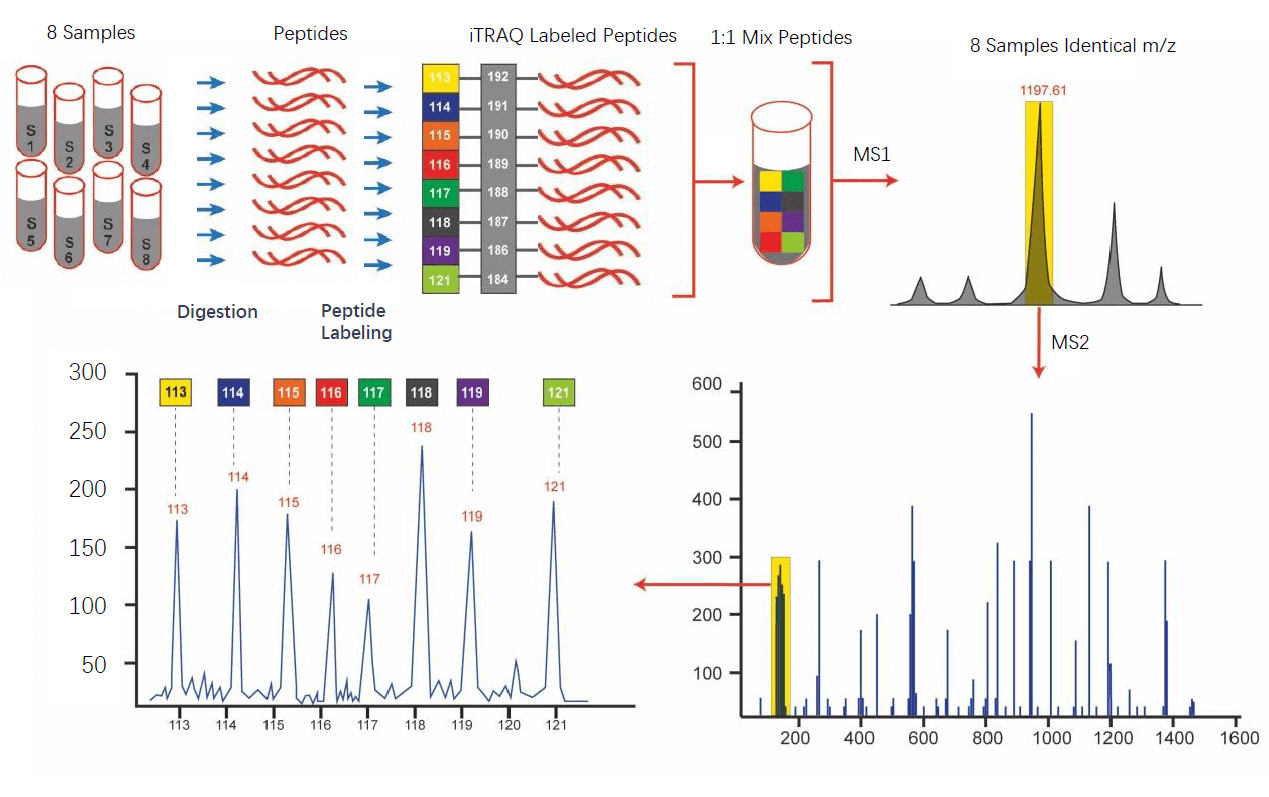
Details
- Sample proteins are reduced and a cysteine blocking agent is added
- Samples are then digested (typically by trypsin) and labelled
- The samples are then pooled and typically passed through a strong cation exchange column to remove excess unbound reagents
- Subjected to tandem-mass-spectrometry
- In the first scan (MS) a given peptide from each sample will produce the same peak
- Then the mass spec puts the sample through a collision cell which further fragments the sample
- This process removes the balance region from the tag, so the same peptide will now have a different m/z from each sample
Tandem mass spectrometry
Fractionation

Identification
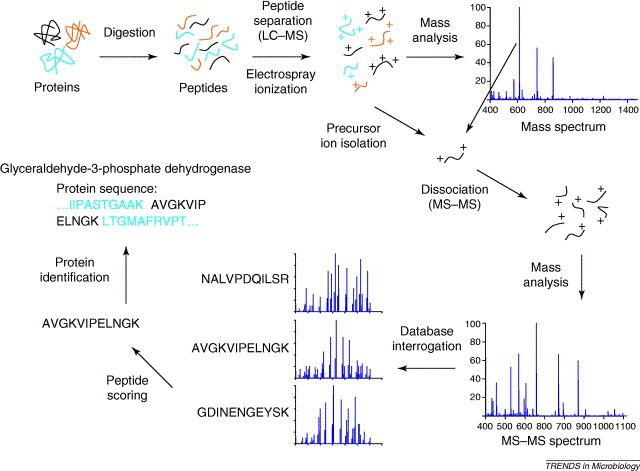
- We can identify peptides from the MS/MS as we know the mass of the elements that make up amino acids
- Therefore we can calculate the mass of the amino acids, and by extension, of peptides by their sequence of AAs.
- So we’d like a database of possible sequences to use for identification
Problem
- This search space is useably large. An 11 AA peptide can have \(2\times 10^{14}\) theoretical sequences
- This didn’t matter for chemists who were only interested in simple molecules, so they could (and often still do) use spectral libraries
- In 1994 a paper was published that pointed out that the search space could be massively reduced by restraining the search to known peptide sequences (identified by genome sequencing) in the organism of interest (as opposed to every theoretical combination) (Eng, McCormack, and Yates, n.d.)
- This combined with limiting to search for possible tryptic cleavage made protein identification viable
- We can then further calculate the theoretical mass of the ions these peptides would generate if they were selected for fragmentation by the instrument
Matching peptide fragments to proteins
#+BEGINNOTES:
- At this point we’ve assigned all of our MS/MS scans to multiple potential peptide fragments
- Each one has a score based on how well the matched fragment matches the theoretical spectra from our database, and an expectation value, which is analogous to a p-value denoting the change that the match is incorrect.
- Once the MS/MS scans are each assigned a peptide sequence that is most likely, the peptides belonging to the same proteins are grouped together, and the protein with the larges number of high scoring peptides is placed at the top of the list
#+ENDNOTES
Details
- At this point we’ve assigned all of our MS/MS scans to multiple potential peptide fragments
- Each one has a score based on how well the matched fragment matches the theoretical spectra from our database, and an expectation value, which is analogous to a p-value denoting the change that the match is incorrect.
- Once the MS/MS scans are each assigned a peptide sequence that is most likely, the peptides belonging to the same proteins are grouped together, and the protein with the larges number of high scoring peptides is placed at the top of the list
Fragmentation ladder
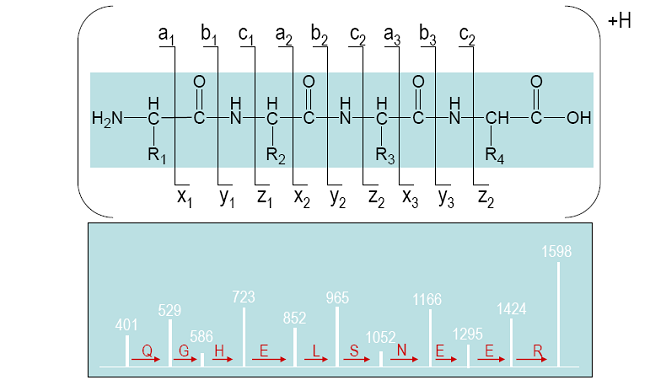
Initial data
| Run | 114 | 115 | 116 | 117 |
|---|---|---|---|---|
| 1 | AIS C Improver @ 2-weeks | AIS C Improver @ 3-months | AIS C Non-Improver @ 2-weeks | AIS C Non-Improver @ 3-months |
| 2 | AIS C Improver @ 2-weeks | AIS C Non-Improver @ 2-weeks | AIS A | AIS D |
C acute run1
C sub-acute
C acute run2
Network plots
Heatmap
Heatmap continued
Cnetplots
Cnetplots continued
Cnetplots continued
Bibliography
Eng, Jimmy K., Ashley L. McCormack, and John R. Yates. n.d. “An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database” 5 (11). American Chemical Society:976–89. https://pubs.acs.org/doi/abs/10.1021/jasms.8b00502.